
Suy tim tâm thất
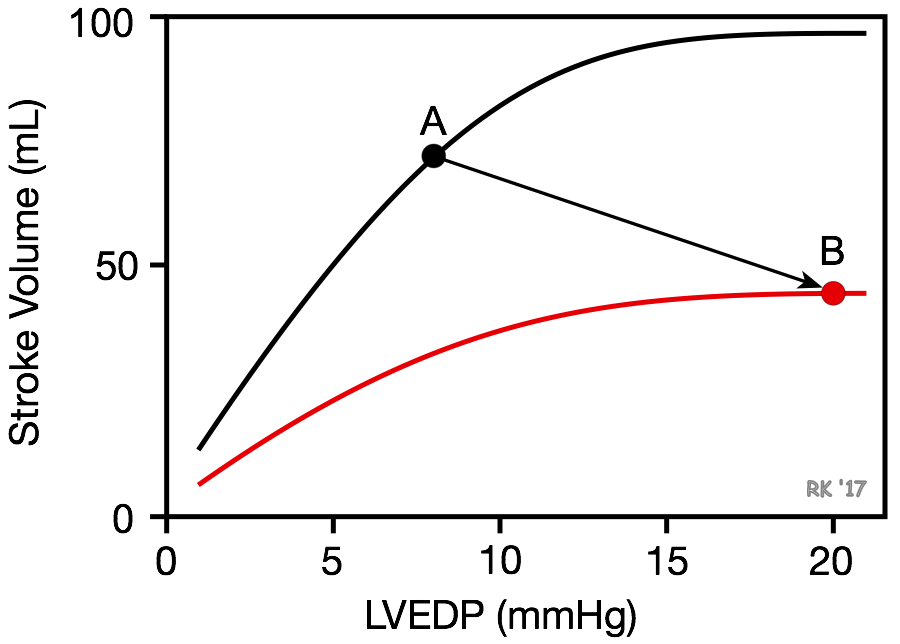
Systolic dysfunction refers to impaired ventricular contraction (loss of inotropy). In chronic heart failure, this is most likely due to changes in the signal transduction mechanisms regulating cardiac excitation-contraction coupling. The loss of cardiac inotropy (i.e., decreased contractility) causes a downward shift in the Frank-Starling curve (red curve in figure). This results in a decrease in stroke volume and a compensatory rise in preload (often measured as ventricular end-diastolic pressure or pulmonary capillary wedge pressure) because of incomplete ventricular emptying, which leads to an increase in ventricular end-diastolic volume and pressure. Therefore, there is a downward/rightward shift in the operating point on the Frank-Starling curve (point A to B). The rise in preload is a compensatory response that activates the Frank-Starling mechanism to help maintain stroke volume despite the loss of inotropy. If preload did not rise, the decline in stroke volume would be even greater for a given loss of inotropy. With systolic dysfunction, there is also an increase in blood volume that contributes to increased ventricular filling and end-diastolic volume and pressure. Ventricular remodeling occurs in chronic failure leading to anatomic dilation of the ventricle.
Effects of systolic dysfunction on ventricular pressure-volume loopThe effects of a loss of intrinsic inotropy on stroke volume, and end-diastolic and end-systolic volumes, are best depicted using ventricular pressure-volume loops (see figure). Loss of intrinsic inotropy decreases the slope of the end-systolic pressure-volume relationship (ESPVR). This leads to an increase in end-systolic volume (red loop). There is also an increase in end-diastolic volume (compensatory increase in preload), but this increase is not as great as the increase in end-systolic volume. Therefore, the net effect is a decrease in stroke volume (shown as a decrease in the width of the pressure-volume loop). Because stroke volume decreases and end-diastolic volume increases, there is a substantial reduction in ejection fraction (EF). In the figure, EF decreases from 58 to 31%. Stroke work (area within loop) is also decreased. Heart failure caused by systolic dysfunction is commonly refered to as heart failure with reduced ejection fraction (HFrEF). Note that with acute systolic dysfunction, there is no change in the end-diastolic pressure-volume relationship (EDPVR). In contrast, with chronic systolic dysfunction the EDPVR relationship would shift downward and to the right as the ventricle responds by remodeling (anatomic dilation), which increases the venticular compliance.

Reduced inotropy effects on force-velocity relationshipThe force-velocity relationship provides insight as to why a loss of contractility causes a reduction in stroke volume (see figure). Briefly, at any given preload and afterload, a loss of inotropy results in a decrease in the shortening velocity of cardiac fibers. Because there is only a finite period of time available for ejection, reduced ejection velocity results in less blood ejected per stroke. The residual volume of blood within the ventricle is increased (increased end-systolic volume) because less blood is ejected.
The reason preload increases as inotropy declines acutely is that the increased end-systolic volume is added to the venous return filling the ventricle. For example, if end-systolic volume is normally 50 ml of blood and it is increased to 80 ml in failure, this extra residual volume added to the incoming venous return leads to an increase in end-diastolic volume and pressure.

An important and deleterious consequence of systolic dysfunction is the rise in end-diastolic pressure. If the left ventricle is involved, then left atrial and pulmonary venous pressures also rise. This can lead to pulmonary congestion and edema. If the right ventricle is in systolic failure, the increase in end-diastolic pressure will be reflected back into the right atrium and systemic venous vasculature. This can lead to peripheral edema and ascites.
Treatment for systolic dysfunction involves the use of inotropic drugs, afterload reducing drugs, venous dilators, and diuretics. Inotropic drugs include digitalis and drugs that stimulate the heart via beta-adrenoceptor activation or inhibition of cAMP-dependent phosphodiesterase. Afterload reducing drugs (e.g., arterial vasodilators) augment ventricular ejection by increasing the velocity of fiber shortening (see force-velocity relationship). Venous dilators and diuretics are used to reduce ventricular preload and venous pressures (pulmonary and systemic) rather than augmenting systolic function directly.
Suy tim tâm trương
Ventricular function is highly dependent upon preload as demonstrated by the Frank-Starling relationship. Therefore, if ventricular filling (preload) is impaired, this will lead to a decrease in stroke volume. The term "diastolic dysfunction" refers to changes in ventricular diastolic properties that have an adverse effect on ventricular filling and stroke volume. About 50% of heart failure patients have diastolic dysfunction, with or without normal systolic function as determined by normal ejection fractions.

Ventricular diastolic dysfunction pressure-volume loopVentricular filling (i.e., end-diastolic volume and hence sarcomere length) depends on the venous return and compliance of the ventricle during diastole. A reduction in ventricular compliance, as occurs in ventricular hypertrophy, increases the slope of the ventricular end-diastolic pressure-volume relationship (EDPVR) and results in less ventricular filling (decreased end-diastolic volume) and a greater end-diastolic pressure (elevated pulmonary capillary wedge pressures) as shown in the figure (red loop). Stroke volume, indicated by the width of the pressure-volume loop, decreases. Depending on the relative change in stroke volume and end-diastolic volume, there may or may not be a small decrease in ejection fraction. The EF of the control loop in the figure is 58% compared to 54% in red loop representing diastolic dysfunction. Heart failure caused by diastolic dysfunction is commonly refered to as heart failure with preserved ejection fraction (HFpEF). Because stroke volume is decreased, there will also be a decrease in ventricular stroke work.
A second mechanism that is non-anatomical, can also contribute to diastolic dysfunction: impaired ventricular relaxation (reduced lusitropy). Near the end of the cycle of excitation-contraction coupling in the myocyte, the sarcoplasmic reticulum actively sequesters Ca++ so that the concentration of Ca++ in the vicinity of troponin-C is reduced allowing the Ca++ to leave its binding sites on the troponin-C and thereby permit disengagement of actin from myosin. This is a necessary step to achieve rapid and complete relaxation of the myocyte. If this mechanism is impaired (e.g., by reduced rate of Ca++ uptake by the sarcoplasmic reticulum), or by other mechanisms that contribute to myocyte relaxation, then the rate and perhaps the extent of relaxation are decreased. This will reduce the rate of ventricular filling, particularly during the phase of rapid filling.
An important and deleterious consequence of diastolic dysfunction is the rise in end-diastolic pressure. If the left ventricle is involved, then left atrial and pulmonary venous pressures will also rise. This can lead to pulmonary congestion and edema. If the right ventricle is in diastolic failure, the increase in end-diastolic pressure will be reflected back into the right atrium and systemic venous vessels. This can lead to peripheral edema and ascites. The rise in venous pressures also occur because of an increase in blood volume due to activation of the renin-angiotensin-aldosterone system, which causes renal retention of sodium and water.
Bệnh lý van
Hẹp van động mạch chủ:
The following describes changes that occur in the left ventricular pressure-volume (PV) loop when there is aortic stenosis. In aortic stenosis (red loop in figure), left ventricular emptying is impaired because of high outflow resistance caused by a reduction in the valve orifice area when it opens. This high outflow resistance causes a large pressure gradient to occur across the aortic valve during ejection, such that the peak systolic pressure within the ventricle is greatly increased. This leads to an increase in ventricular wall stress (afterload), a decrease in stroke volume, and an increase in end-systolic volume. Stroke volume (width of PV loop) decreases because the velocity of fiber shortening is decreased by the increased afterload (see force-velocity relationship). Because end-systolic volume is elevated, the excess residual volume added to the incoming venous return causes the end-diastolic volume to increase. This increases preload and activates the Frank-Starling mechanism to increase the force of contraction to help the ventricle overcome, in part, the increased outflow resistance. In mild aortic stenosis, this can be adequate to maintain normal stroke volume, but in moderate stenosis (as shown in the figure) or severe stenosis, the stroke volume may fall considerably because the end-systolic volume increases substantially more than the end-diastolic volume increases. The fall in stroke volume can lead to a reduction in arterial pressure. Stroke volume falls even further if the ventricle begins to exhibit systolic and diastolic dysfunction. Compensatory increases in end-diastolic volume will be limited by ventricular hypertrophy that occurs due to the chronic increase in afterload. This hypertrophy can lead to a large increase in end-diastolic pressure that is associated with reduced end-diastolic volumes because the increased stiffness of the ventricle prevents normal ventricular filling.

The changes described above and shown in the figure do not include cardiac and systemic compensatory mechanisms (e.g., systemic vasoconstriction, increased blood volume, and increased heart rate and inotropy) that attempt to maintain cardiac output and arterial pressure. Therefore, the red loop depicted in the figure only represents what may occur under a given set of conditions.
Hẹp van hai lá:
The following describes changes that occur in the left ventricular pressure-volume (PV) loop when there is mitral stenosis. Mitral stenosis (red PV loop in figure) impairs left ventricular filling so that there is a decrease in end-diastolic volume (preload). This leads to a decrease in stroke volume (reduced width of PV loop) by the Frank-Starling mechanism and a fall in cardiac output. Reduced ventricular filling and reduced aortic pressure decrease ventricular wall stress (afterload), which may result in a small decrease in ventricular end-systolic volume; however, this is not sufficient to offset the reduction in end-diastolic volume. Therefore, because end-diastolic volume decreases more than end-systolic volume decreases, the stroke volume (shown as the width of the loop) decreases.
The changes described above and shown in the figure do not include cardiac and systemic compensatory mechanisms (e.g., systemic vasoconstriction, increased blood volume, and increased heart rate and inotropy) that attempt to maintain cardiac output and arterial pressure. Therefore, the red loop depicted in the figure only represents what may occur under a given set of conditions.

Sự chảy ngược van hai lá (Hở van hai lá)
The following describes changes that occur in the left ventricular pressure-volume loop when there is mitral regurgitation. In mitral valve regurgitation (red pressure-volume loop in figure), as the left ventricle contracts, blood is not only ejected into the aorta but also back up into the left atrium. This causes left atrial volume and pressure to increase during ventricular systole. Note in the pressure-volume loop that there is no true isovolumetric contraction phase (no vertical line between mitral valve closure and aortic valve opening) because blood begins to flow across the mitral valve and back into the atrium before the aortic valve opens as soon as ventricular pressure exceeds left atrial pressure. Because of mitral regurgitation, the afterload on the ventricle is reduced (total outflow resistance is reduced) so that end-systolic volume can be smaller than normal; however, end-systolic volume can increase if the heart also goes into systolic failure. There is no true isovolumetric relaxation (vertical lines between aortic valve closure and mitral valve opening) because when the aortic valve closes and the ventricle begins to relax, the mitral valve is not completely closed so blood continues to flow back into the left atrium (therefore further decreasing ventricular volume) as long as intraventricular pressure is greater than left atrial pressure. During ventricular diastolic filling, the elevated pressure within the left atrium is transmitted to the left ventricle during filling so that left ventricular end-diastolic volume (and pressure) increases. Ventricular end-diastolic volume is also increased because in chronic mitral regurgitation the ventricle anatomically dilates (remodels) so that ventricular compliance is elevated. Increased end-diastolic volume would cause wall stress (afterload) to increase if it were not for the reduced outflow resistance because of mitral regurgitation that tends to decrease afterload during ejection because of reduced pressure development by the ventricle. The net effect of these changes is that the width of the pressure-volume loop is increased (i.e., ventricular stroke volume is increased); however, ejection into the aorta (forward flow) is reduced. The increased ventricular "stroke volume" (measured as the end-diastolic minus the end-systolic volume) in this case includes the volume of blood ejected into the aorta as well as the volume ejected back into the left atrium.

The changes just described do not include cardiac and systemic compensatory mechanisms (e.g., systemic vasoconstriction, increased blood volume, and increased heart rate and inotropy) that attempt to maintain cardiac output and arterial pressure. Therefore, the red loop depicted in the figure only represents what may occur under a given set of conditions.
Hở van động mạch chủ
The following describes changes that occur in the left ventricular pressure-volume loop when there is aortic regurgitation. In aortic valve regurgitation (red loop in figure), the aortic valve does not close completely at the end of systolic ejection. As the ventricle relaxes during diastole, blood flows from the aorta back into the ventricle so the ventricle immediately begins to fill from the aorta. Therefore, there is no true phase of isovolumetric relaxation (no vertical line between aortic valve closure and mitral valve opening) because as the ventricle relaxes, even before the mitral valve opens, blood is entering the ventricle from the aorta thereby increasing ventricular volume. Once the mitral valve opens, filling occurs from the left atrium; however, blood continues to flow from the aorta into the ventricle throughout diastole because aortic pressure is higher than ventricular pressure during diastole. This greatly enhances ventricular filling so that end-diastolic volume is increased as shown in the pressure-volume loop. Ventricular end-diastolic volume is also increased because in chronic aortic regurgitation the ventricle anatomically dilates (remodels) so that ventricular compliance is elevated.

When the ventricle begins to contract and develop pressure, blood is still entering the ventricle from the aorta because aortic pressure is higher than ventricular pressure; therefore, there is no true isovolumetric contraction because volume continues to increase. Once the ventricular pressure exceeds the aortic diastolic pressure, the ventricle then begins to eject blood into the aorta. The increased end-diastolic volume (increased preload) activates the Frank-Starling mechanism to increase the force of contraction, ventricular peak (systolic) pressure, and stroke volume (as shown by the increased width of the pressure-volume loop). As long as the ventricle is not in failure, end-systolic volume may only be increased a small amount (as shown in figure) due to the increased afterload (ventricular wall stress). If the ventricle goes into systolic failure, then end-systolic volume will increase by a large amount and the peak systolic pressure and stroke volume (net forward flow into aorta) will fall.
The changes just described do not include cardiac and systemic compensatory mechanisms (e.g., systemic vasoconstriction, increased blood volume, and increased heart rate and inotropy) that attempt to maintain cardiac output and arterial pressure. Therefore, the red loop depicted in the figure only represents what may occur under a given set of conditions.
Post a Comment